
SCD is an inherited disease. It follows an autosomal recessive pattern, meaning a child will not inherit the disease unless both parents pass down a defective copy of the gene. People who inherit one good copy of the gene and one mutated copy are carriers. They are clinically normal, but they can still pass the defective gene to their children (Fig. 2).
SCD is much more common in people of African and Mediterranean descent. It is also seen in people from South and Central America, the Caribbean, and the Middle East (Fig. 3).
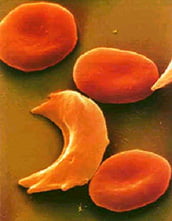 Figure 1: SCD morphology; normal, round red blood cells and sickle-shaped red blood cell
Figure 1: SCD morphology; normal, round red blood cells and sickle-shaped red blood cell Figure 2: SCD probability
Figure 2: SCD probability Figure 3: Global distributio of SCD
Figure 3: Global distributio of SCDSCD prevents oxygen from reaching the important organs and without oxygen, the cells that make up these organs begin to die. As a result, SCD patients often experience frequent infections. Because the red blood cells of patients with sickle cell disease don't live as long as healthy red blood cells (10-20 days compared to 120 days), people with this disease often have low red blood cell counts (anemia), which is why this disease is commonly referred to as “sickle cell anemia.”
When sickle-shaped red blood cells get stuck in blood vessels, it can cause episodes of pain known as “crises.” Other symptoms include delayed growth, stroke, and jaundice (yellowish hue to the skin and eyes because of liver damage). Ischemic stroke will occur in over 10% of children with SCD by the age of 20. In the past, people with sickle cell disease often died between the ages of 20 and 40. Thanks to better care, today people with SCD can live to the age of 50 and beyond.
SCD can be diagnosed by a simple blood test. Babies and young children with SCD take penicillin daily to prevent potentially deadly infections. They also take folic acid, which helps build new red blood cells. People with SCD are advised to get plenty of rest, drink lots of water, and avoid too much physical activity. Blood transfusions to provide healthy red blood cells are a common treatment. People with more severe cases of the disease can be treated with a bone marrow transplant.
SCD, Transcranial Doppler (TCD) & Stroke
TCD is used to evaluate cerebral arterial hemodynamics, and it plays an important role in screening and providing a prognosis for SCD patients.
The stroke prevention trials in sickle cell anemia (STOP trials I and II) used TCD to screen and identify SCD patients who were at the greatest risk of ischemic stroke. The trial found that patients with increased velocity in the distal internal carotid artery (dICA) and the proximal middle cerebral artery (pMCA) had the greatest risk for stroke. Once these patients were identified, confirmed, and treated with prophylactic blood transfusion, the STOP trial demonstrated a more than 90% reduction in the risk of stroke. As of November 2012, the STOP trial results were integrated in the Society of Vascular Ultrasound (SVU) guidelines for using TCD in sickle cell anemia pediatric patients. The guidelines state that a TCD examination should be performed once or twice a year for SCD patients starting at the age of 2.
Mean flow velocity in the dICA and pMCA is greater than 200 cm/sec in two consecutive examinations indicates a high risk of stroke, and the patient is treated with prophylactic blood transfusion.
Digi-Lite TCD
Digi-Lite TCD is noninvasive, does not require sedation, and is easily repeatable, making it a safe, comfortable choice for SCD-patient screening.
Resources
Some information appears on quizlet.com/195749550/genetics-concepts-flash-cards.


YOUR SAFE CHOICE
Get an online demo